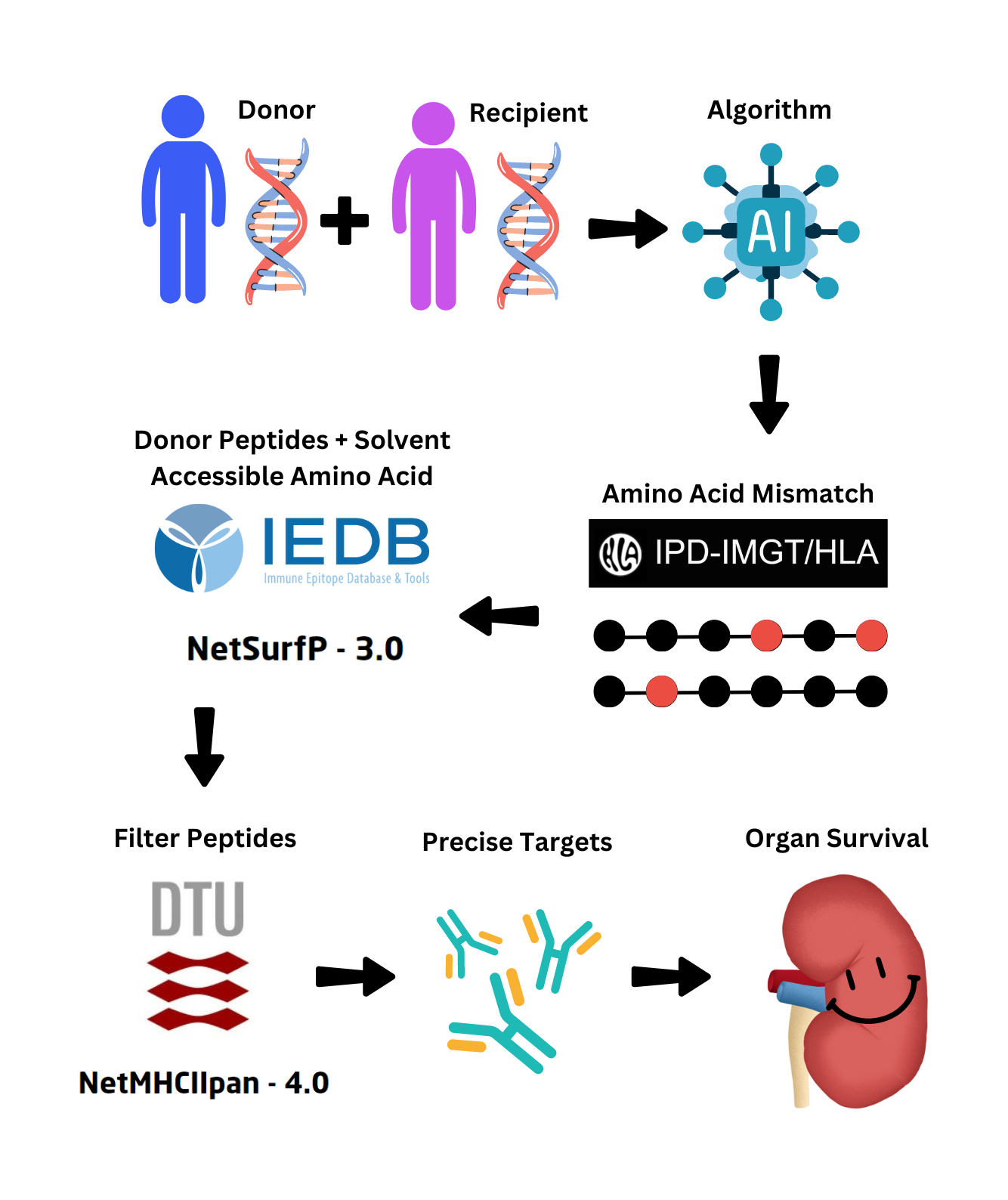
Data Collection and Preprocessing:
HLA Protein Sequences. The Immuno-Polymorphism Database (IPD-IMGT/HLA)bversion 3.55.0 from the European Bioinformatics Institute (EBI) was accessed
through the database’s public FTP site hosted by the EBI. The database provides a central repository for sequences of HLA alleles,
including the protein sequences in the FASTA format. HLA allele sequences were filtered to only include the commonly typed HLA loci:
HLA‐A, ‐B, ‐C, ‐DRB1, ‐DRB3, ‐DRB4, ‐DRB5, −DQA1, ‐DQB1, ‐DPA1 and ‐DPB1 (Hamed et al., 2018). The alleles were converted into field
type two resolutions, as higher resolution typing does not affect the amino acid sequence of the protein (Kramer et al., 2020).
Study Cohorts. The STAR files were obtained by the United Network for Organ Sharing (U.N.O.S.), which include donor and recipient
transplant data dated back to 1987. The large dataset was processed, resulting in a small, manageable dataset with living kidney
transplantations. The dataset contains past donor and recipient HLA alleles along with the rejection outcome. Chronic rejection was
defined as rejection episodes that occur at least one year after the transplant (Vaillant & Mohseni, 2023).
HLA-Epi is another model that calculated the epitopic mismatch load between potential recipient-donor pairs. The HLA-Epi dataset contains
donor and recipient HLA alleles along with their calculated compatibility scores (Geffard et al., 2022). Even though the model focuses on
direct allorecognition, the compatibility scores can be used to validate the proposed model’s performance through regression models.
Additionally, they have scores calculated by the PIRCHE-II model for the same donor and recipient alleles. The PIRCHE-II model is
another algorithm to predict indirectly recognizable HLA epitopes (Geneugelijk & Spierings, 2020). The PIRCHE-II model does not consider
solvent-accessible mismatches. Therefore, the scores in the HLA-Epi dataset can be used to compare the performance of the proposed model
with competitor models.
Bioinformatics Servers:
Bioinformatic servers were used to analyze and compare the amino
acid sequences of donor and recipient HLA alleles. NetSurfP from the
Danmarks Tekniske Universitet (DTU Health Tech) was used to predict the surface accessibility of individual amino acids in an
amino acid sequence.
Additionally, NetMHCIIpan from DTU Health Tech was used to predict the binding affinity and eluted ligand of donor HLA peptides
to recipient
HLA class II alleles.
Software and Software Packages:
Google Colaboratory was used to code the machine learning models, as it is a
hosted Jupyter Notebook to write and execute Python code through the browser. Microsoft Excel was used to format the data in a table format
to make it easier to upload as a data frame into Google Collab. The HLA Epitope Mismatch Algorithm (HLA-EMMA) was used to validate amino
acid mismatch results. Python libraries such as “Pandas” were used to import Excel data files, and “NumPy” was used to support the
large arrays in the data files. Additionally, “MatplotLib” was used to visualize data, and “Seaborn” was used to create a confusion
matrix. Lastly, the Statistical Analysis System (SAS) software will be used to convert the U.N.O.S. data files into a readable Excel file.
Modified Needleman-Wunsch Algorithm:
The IPD/IMGT-HLA database has allele sequences in different lengths. However,
to find the amino acid mismatches, the sequences must be of equal length to be vertically aligned. Therefore, a modified Needleman-Wunsch
algorithm was used to make the sequences have equal lengths. The Needleman-Wunsch algorithm is a common global alignment method that uses
a scoring matrix and dynamic programming to find the optimal alignment between two sequences (Mittal, 2024). The traditional algorithm adds gaps
between the protein sequences, representing the evolutionary changes between the two sequences. The gaps attempt to optimize the alignment score
and reveal any mutations, insertions, or deletions that may have occurred over time (NandiniUmbarkar, 2020). However, to find the amino acid
mismatches between the donor and recipient FASTA sequences, there should not be any additional modifications to the sequence. Therefore,
the model uses a similar scoring system but has a very high gap penalty. The gap penalty is a negative score that is added to the
score any time a gap is inserted in the sequences (Mount, 2008). By having a high negative gap penalty, the overall score will significantly
decrease. To have a high alignment score, the sequences will not be modified.
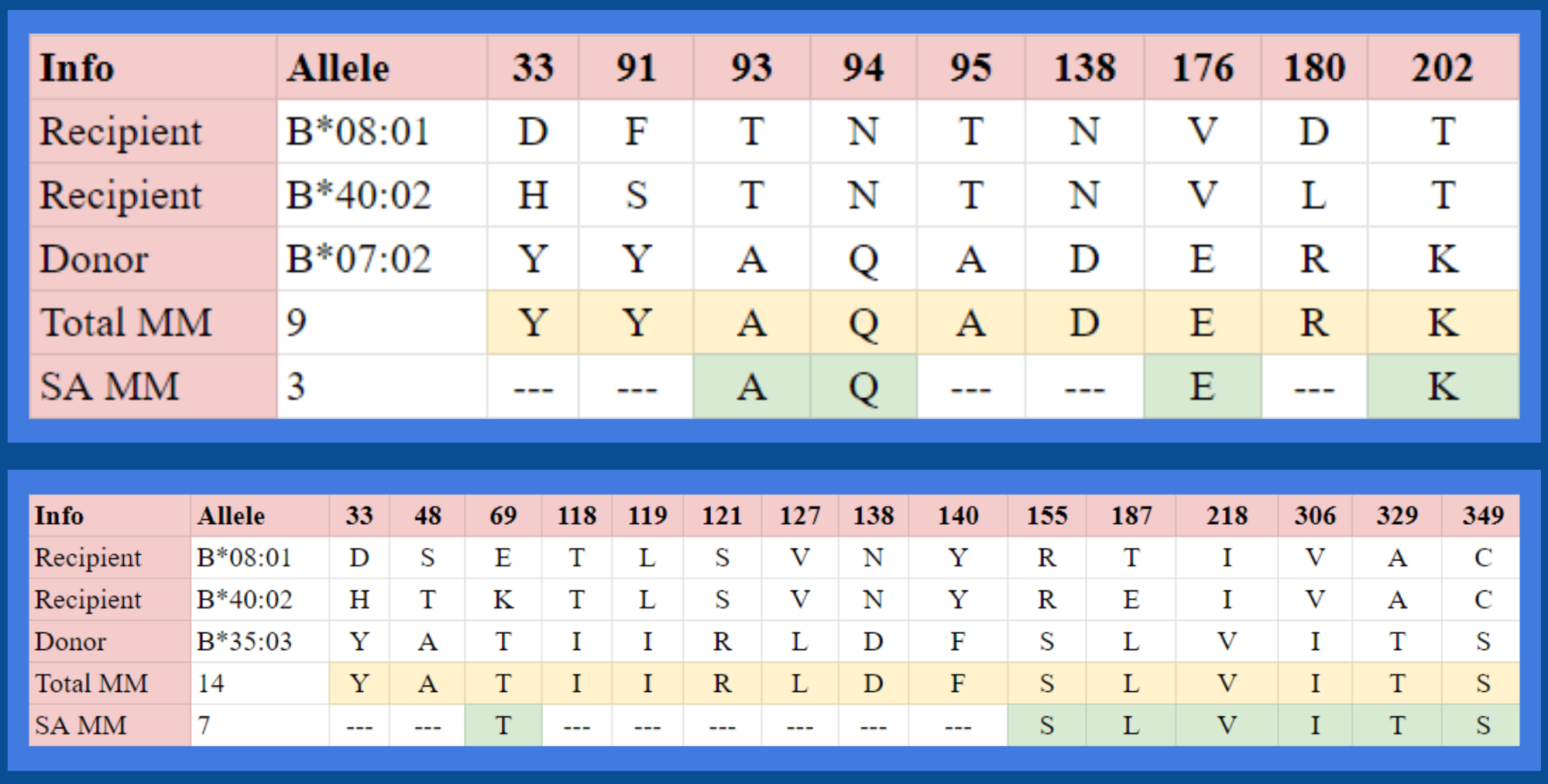
Finding Solvent-Accessible Amino Acid Mismatches
Solvent-accessible amino acids are amino acids in a protein that
are exposed to the solvent surrounding the protein. These amino acids have a
much higher chance of being recognized by T-cells. Therefore, NetSurfP was used to predict the solvent accessibility for each
amino acid in the donor alleles, and the
solvent-accessible amino acids which also contained amino acid mismatches were stored for peptide analysis.
Generating Donor-Derived Peptide Chains
NetMHCIIpan was used to generate donor-derived peptides that were
15 amino acids in length. The binding affinity and
eluted ligand were found for all generated peptides to find the strongest peptides. The donor alleles were used
for peptide sequence generation, and the molecules were input as the recipient MHC class II molecules.
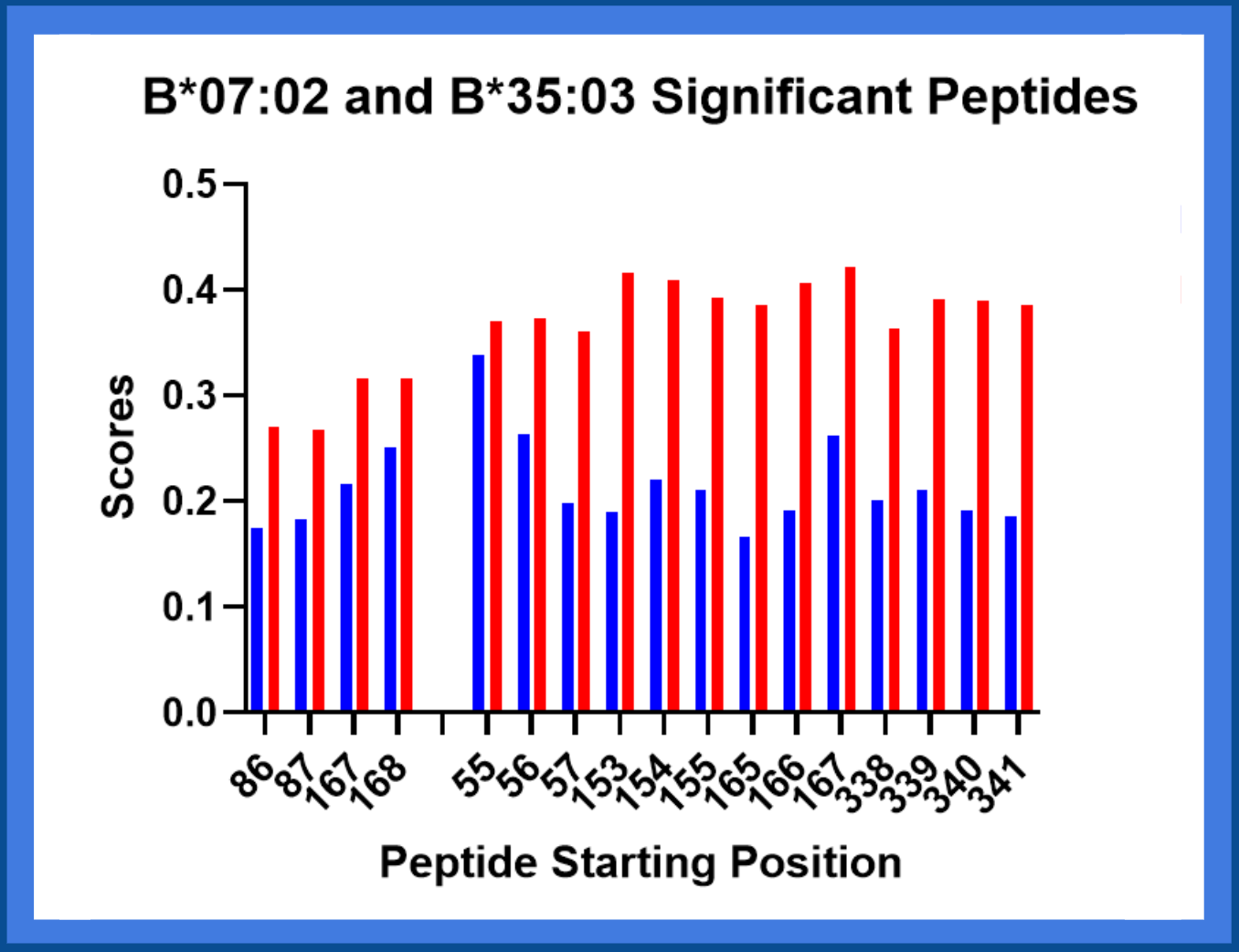
Filtering Peptides With Binding Affinity and Eluted Ligand
The eluted ligand score is the likelihood of a peptide being an
MHC ligand, while binding affinity is the strength of attraction between the peptide and
the molecule (Wongklaew et al., 2024). NetMHCIIpan reports the strongest binding peptide sequences to each MHC class II molecule.
Out of those, the
peptides containing the solvent-accessible amino acid mismatches were stored as the most significant peptides that may cause
rejection.
Machine-Learning Model Training and Testing:
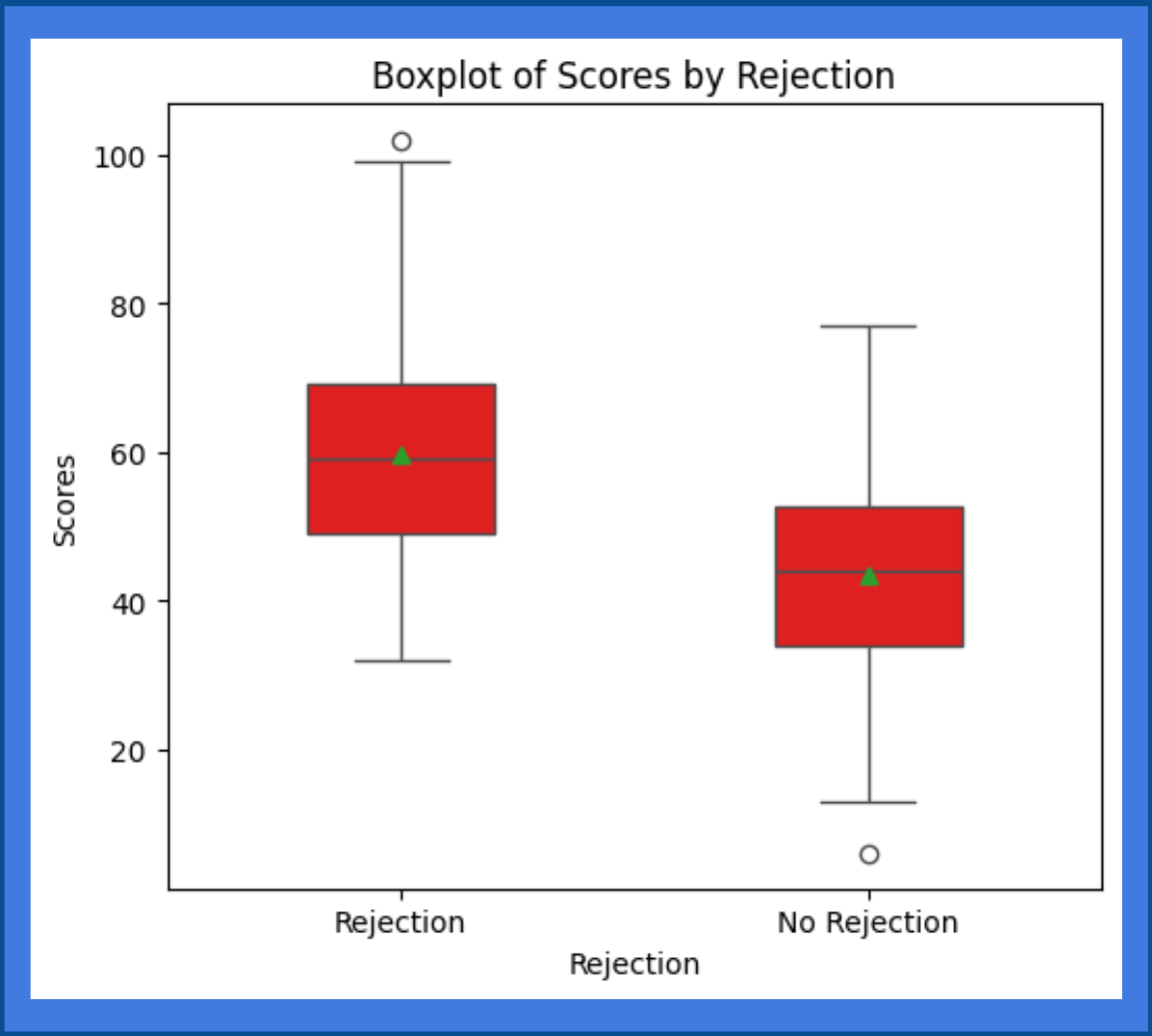
After the model is completed, the HLA-Epi data will be used to create regression
models between the predicted compatibility score and the true compatibility score. The donor and recipient samples be run through the model,
and the predicted scores will be recorded. Then, the true scores of the respective samples will be matched with the predicted score from the
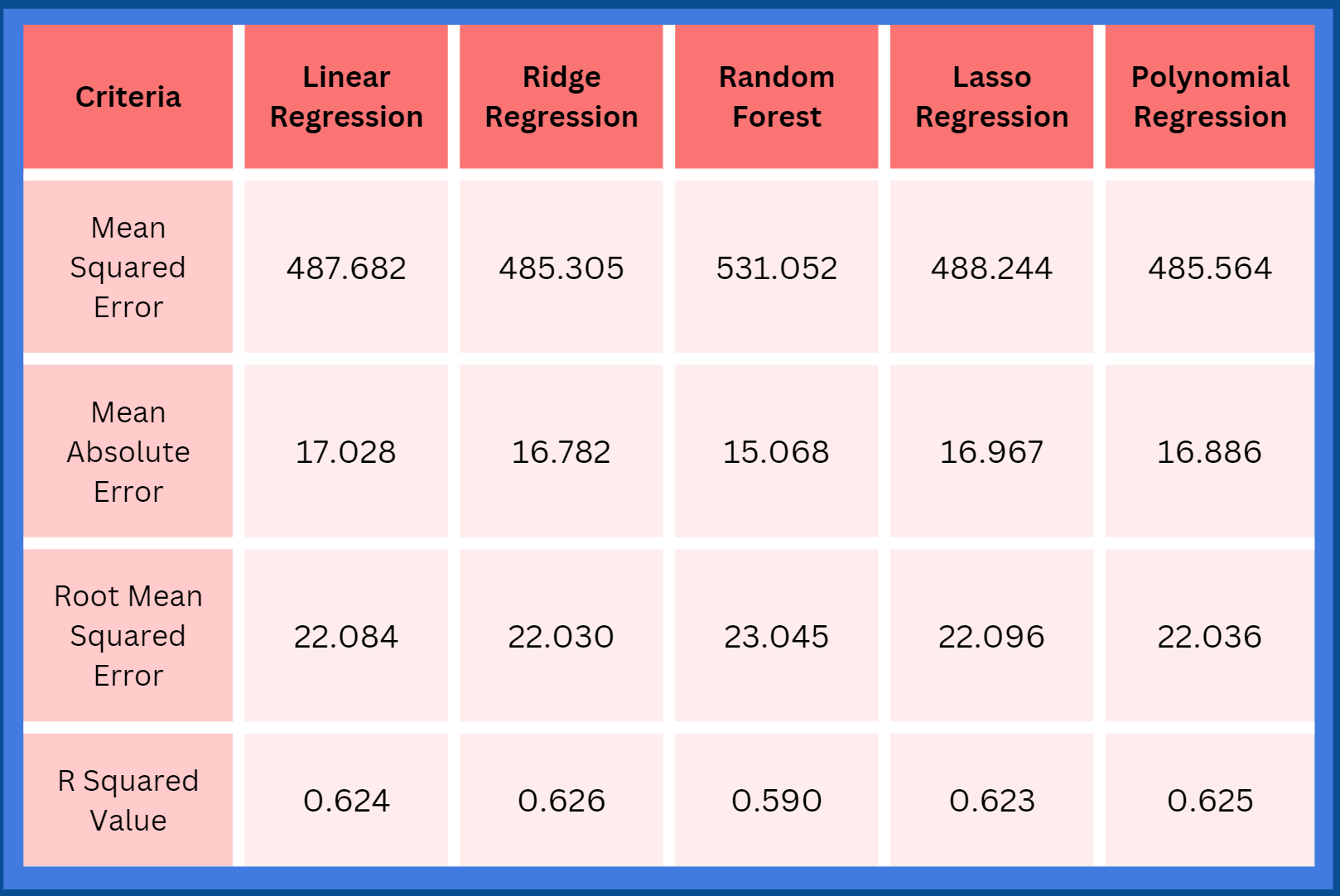
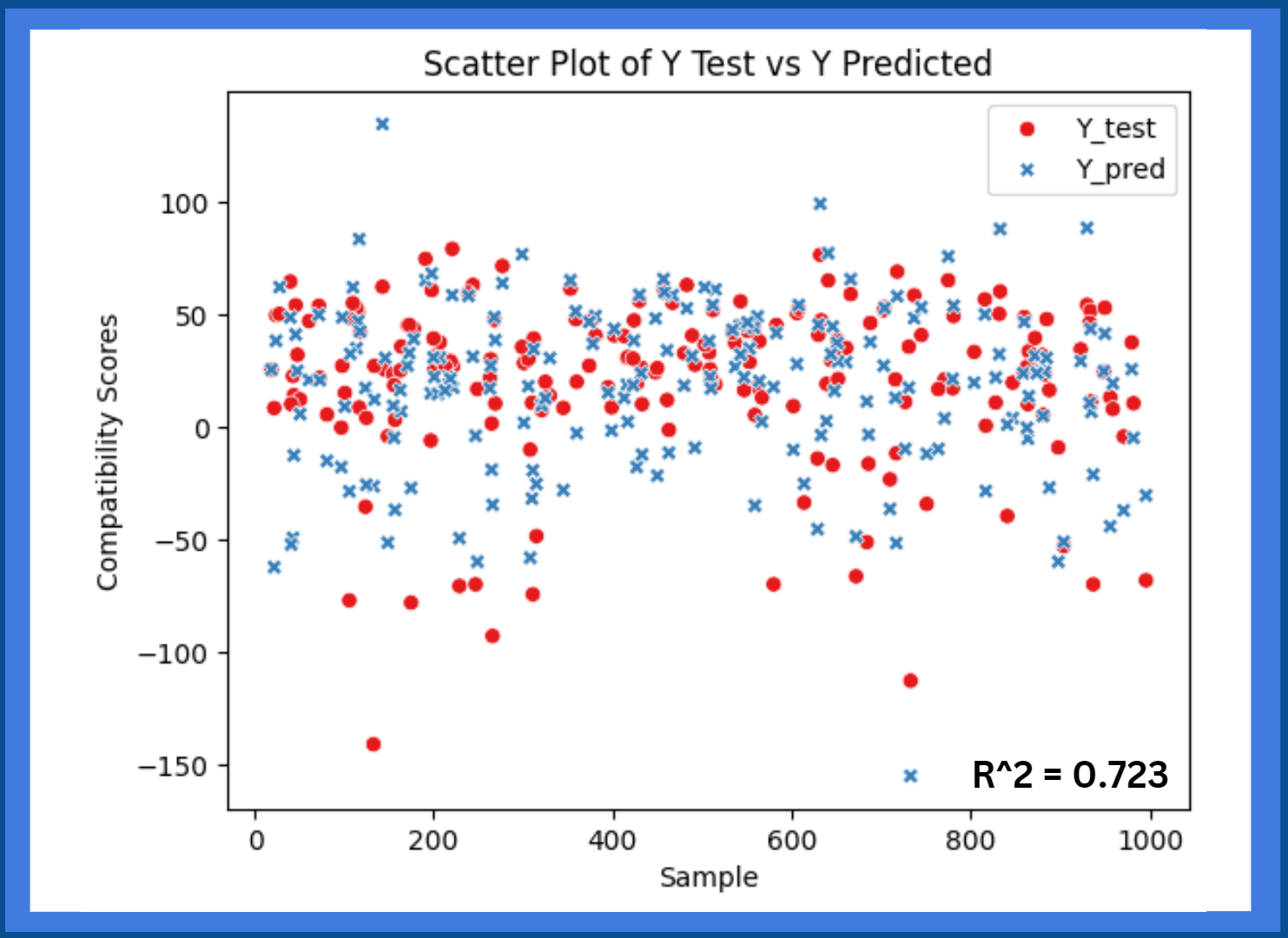
model. Regression models will be made to validate the model’s ability to accurately predict a score for a sample on a scale. The model
will be improved until it reaches an accuracy of at least 70% or greater. If needed, feature selection algorithms such as random forest will
be used to find the most influential HLA alleles, which can improve the accuracy of the regression models.